10X Example
Last updated: 2018-09-14
workflowr checks: (Click a bullet for more information)-
✔ R Markdown file: up-to-date
Great! Since the R Markdown file has been committed to the Git repository, you know the exact version of the code that produced these results.
-
✔ Environment: empty
Great job! The global environment was empty. Objects defined in the global environment can affect the analysis in your R Markdown file in unknown ways. For reproduciblity it’s best to always run the code in an empty environment.
-
✔ Seed:
set.seed(20180618)The command
set.seed(20180618)was run prior to running the code in the R Markdown file. Setting a seed ensures that any results that rely on randomness, e.g. subsampling or permutations, are reproducible. -
✔ Session information: recorded
Great job! Recording the operating system, R version, and package versions is critical for reproducibility.
-
Great! You are using Git for version control. Tracking code development and connecting the code version to the results is critical for reproducibility. The version displayed above was the version of the Git repository at the time these results were generated.✔ Repository version: 86bd1c0
Note that you need to be careful to ensure that all relevant files for the analysis have been committed to Git prior to generating the results (you can usewflow_publishorwflow_git_commit). workflowr only checks the R Markdown file, but you know if there are other scripts or data files that it depends on. Below is the status of the Git repository when the results were generated:
Note that any generated files, e.g. HTML, png, CSS, etc., are not included in this status report because it is ok for generated content to have uncommitted changes.Ignored files: Ignored: .Rhistory Ignored: .Rproj.user/ Ignored: R/.Rhistory Ignored: analysis/.Rhistory Ignored: analysis/pipeline/.Rhistory Untracked files: Untracked: ..gif Untracked: .DS_Store Untracked: R/.DS_Store Untracked: analysis/.DS_Store Untracked: analysis/normalization_test.R Untracked: analysis/pipeline/0_dropseq/ Untracked: analysis/pipeline/1_10X/ Untracked: analysis/pipeline/2_zeisel/ Untracked: analysis/pipeline/3_smallsets/ Untracked: analysis/slsl_10x.Rdata Untracked: analysis/slsl_dropseq.Rdata Untracked: analysis/writeup/bibliography.bib Untracked: analysis/writeup/draft1.aux Untracked: analysis/writeup/draft1.bbl Untracked: analysis/writeup/draft1.blg Untracked: analysis/writeup/draft1.log Untracked: analysis/writeup/draft1.out Untracked: analysis/writeup/draft1.pdf Untracked: analysis/writeup/draft1.synctex.gz Untracked: analysis/writeup/draft1.tex Untracked: analysis/writeup/jabbrv-ltwa-all.ldf Untracked: analysis/writeup/jabbrv-ltwa-en.ldf Untracked: analysis/writeup/jabbrv.sty Untracked: analysis/writeup/naturemag-doi.bst Untracked: analysis/writeup/wlscirep.cls Untracked: data/unnecessary_in_building/ Untracked: docs/figure/10x_labeled.Rmd/ Untracked: docs/figure/example_10x.Rmd/.DS_Store Untracked: docs/figure/small.Rmd/ Untracked: dropseq_heatmap.pdf Untracked: man/correct_detection_rate.Rd Unstaged changes: Modified: NAMESPACE Modified: R/SLSL.R Modified: R/correct_detection_rate.R Modified: R/find_markers.R Modified: analysis/pipeline/.DS_Store Modified: analysis/writeup/.DS_Store Modified: data/.DS_Store Modified: docs/figure/.DS_Store Deleted: man/correct.detection.rate.Rd
Expand here to see past versions:
| File | Version | Author | Date | Message |
|---|---|---|---|---|
| Rmd | 86bd1c0 | tk382 | 2018-09-14 | wflow_publish(c(“analysis/_site.yml“,”analysis/large.Rmd“,”analysis/small.Rmd“), republish |
| html | 32ed2c0 | tk382 | 2018-09-13 | small edits |
| Rmd | 047143d | tk382 | 2018-09-12 | large set pipeline |
| html | 047143d | tk382 | 2018-09-12 | large set pipeline |
| Rmd | 03a7f75 | tk382 | 2018-09-12 | notation change, adding visualization, general code structure |
Load Package
Read Data : PBMC 27K
Read data and keep the gene names separately.
orig = readMM('data/unnecessary_in_building/pbmc3k/matrix.mtx')
orig_genenames = read.table('data/unnecessary_in_building/pbmc3k/genes.tsv',
stringsAsFactors = FALSE)$V2Quality Control and Cell Filtering
First, “explore.data” function shows the distribution of the number of UNI, detection rate, and the proportion of mitochondrial genes for each cell. Then users can decide on the cut-offs to filter the cells using “cell.filter” function, which returns the indices of cells that satisfy the input criteria.
summary = explore_data(orig, orig_genenames)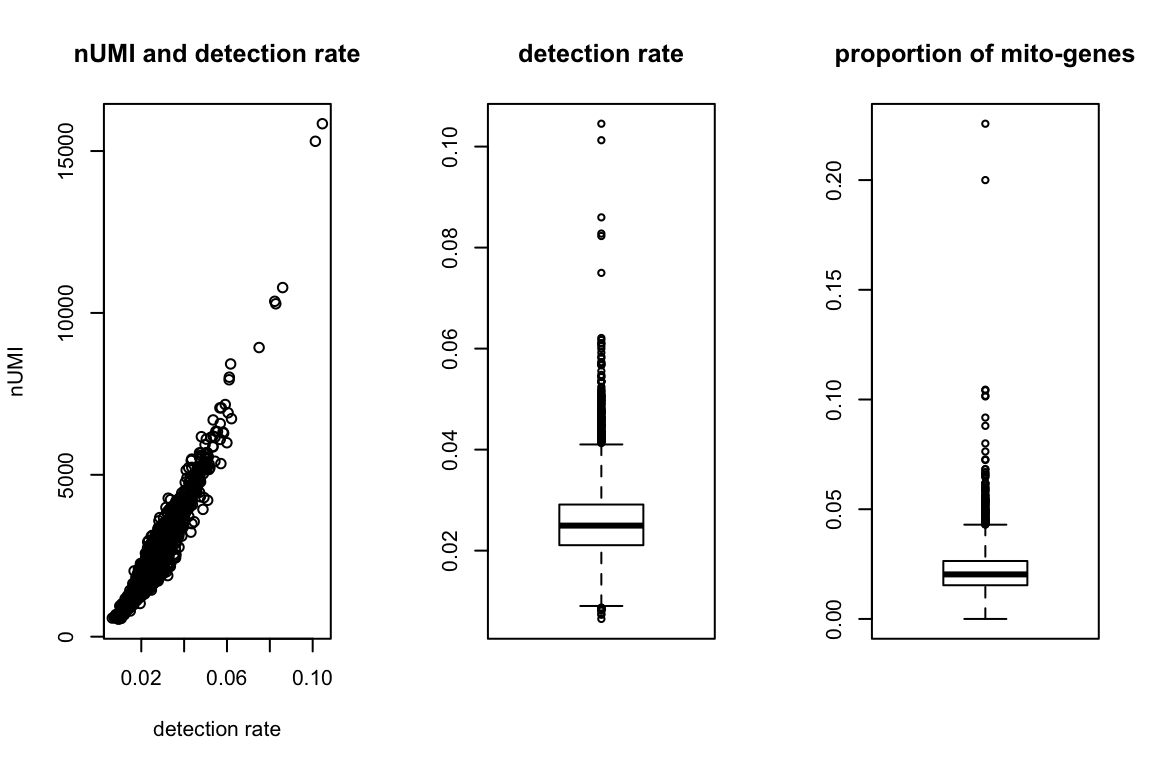
ind = cell_filter(summary,
nUMI.thresh = c(0,10000),
det.rate.thresh = c(0.02, 0.067),
percent.mito.thresh = c(0,0.1))
tmpX = orig[,ind]Gene Filtering
Next find variable genes using normalized dispersion. First, “plot.dispersion” function shows the distribution log of normalized dispersion (variance to mean ratio) against the gene means. The convention is to normalize the dispersion by converting them to z-scores, but one can also use median and MAD (median absolute deviation) which is less sensitive to outliers. Based on the scatterplot, users can decide on the cut-offs for the mean expression value and the dispersion to use “gene.filter” function. “gene.filter” function returns the new expression level matrix of filtered genes and the corresponding gene names.
disp = plot_dispersion(X = tmpX,
genenames = orig_genenames,
bins=NA,
median = FALSE,
outliers.mean.thresh = c(30,Inf),
outliers.vmr.thresh = c(3,Inf))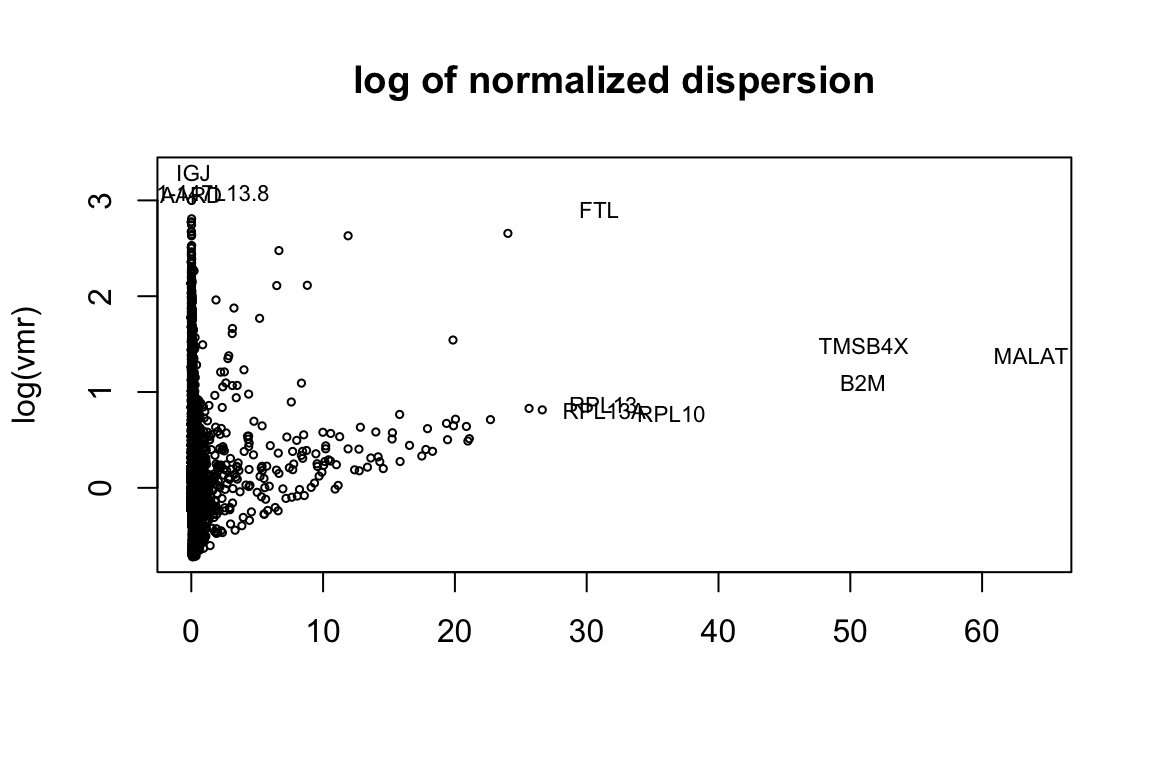
X = gene_filter(tmpX, orig_genenames, disp,
mean.thresh=c(0.001, Inf),
dispersion.thresh = c(0.5, Inf))
genenames = X$genenames
X = X$XUMI Normalization
Use quantile-normalization to make the distribution of each cell the same. “quantile.normalize” function also performs the log transformation.
X = quantile_normalize(as.matrix(X))Run SLSL
Run the clustering algorithm SLSL based on the filtered matrix. It automatically plots the final tSNE plot based on the Laplacian matrix.
out = SLSL(X, verbose=FALSE)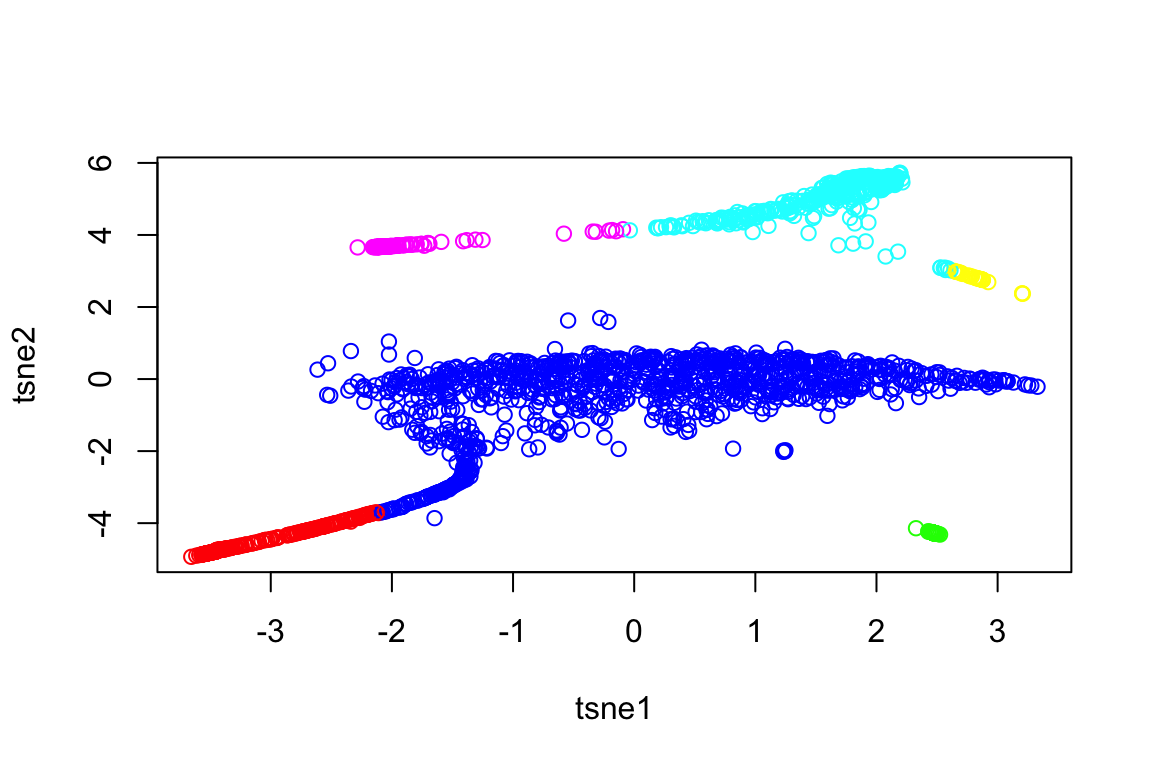
Differentially Expressed Genes
Using Kruskal test, we order the p-values to find the top differentially expressed genes. Below we present 6.
degenes = de_genes(X, genenames, out$result, top.n=100, plot=12)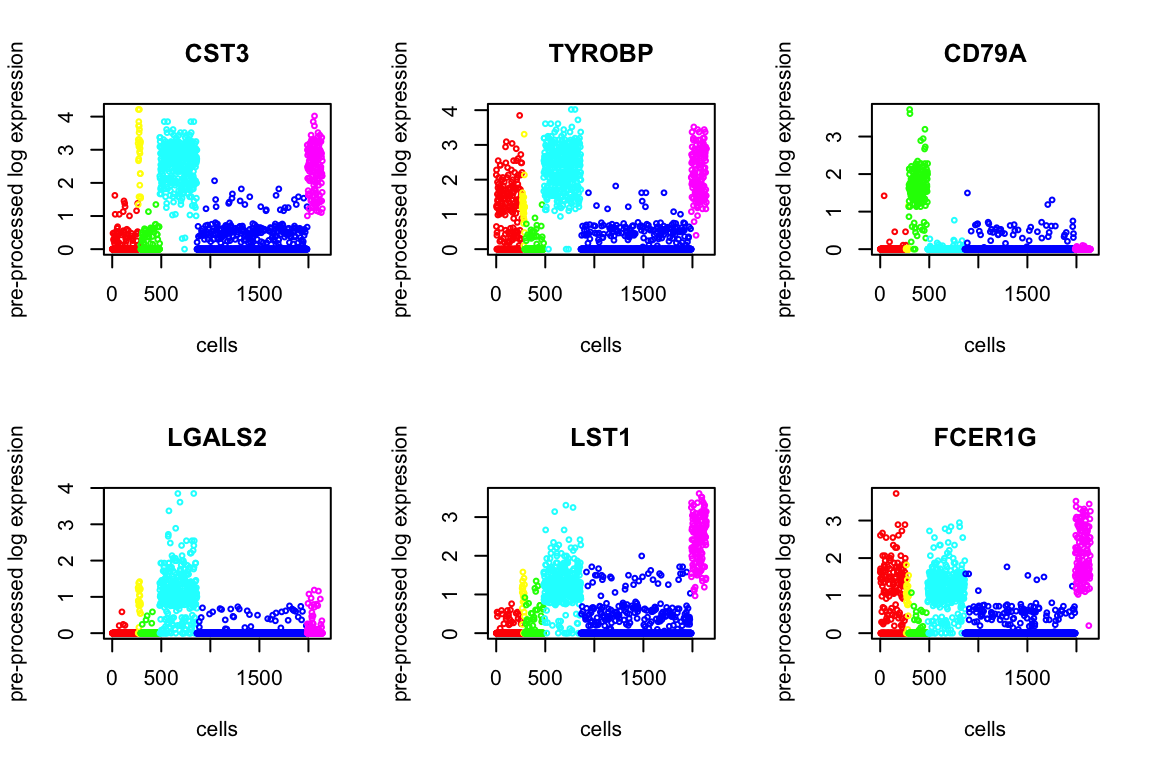
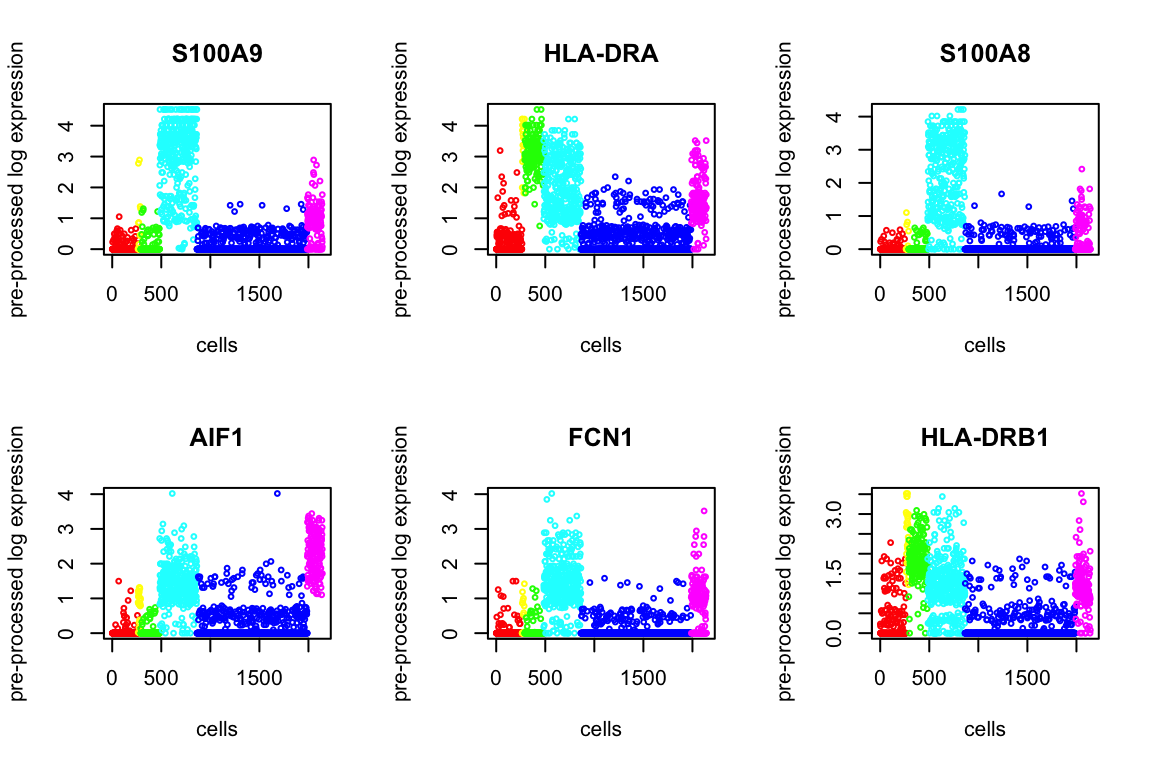
head(degenes) de_genes log10p
1 CST3 Inf
2 TYROBP Inf
3 CD79A Inf
4 LGALS2 Inf
5 LST1 322.2270
6 FCER1G 314.1417Markers for Each Cluster
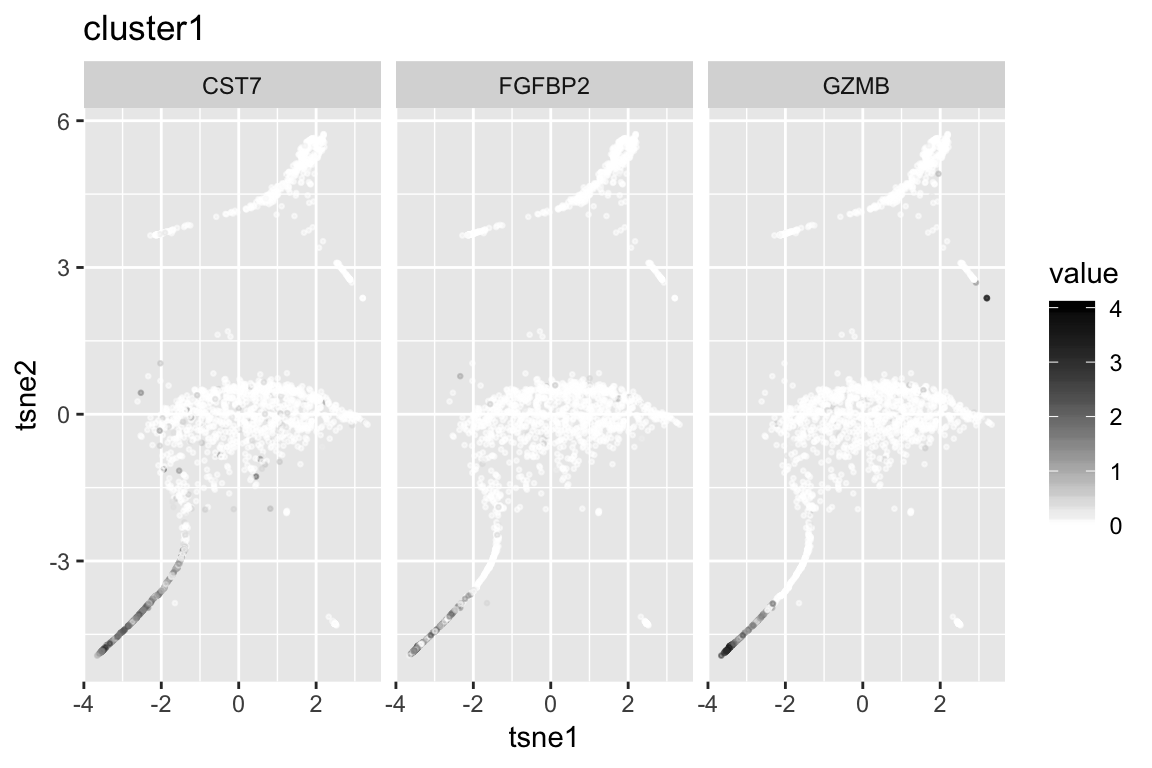
clust1_genenames clust1_log10p
1 GZMB 280.5690
2 FGFBP2 249.4204
3 CST7 239.9105
4 PRF1 220.0278
5 NKG7 213.0282
6 CCL4 167.3846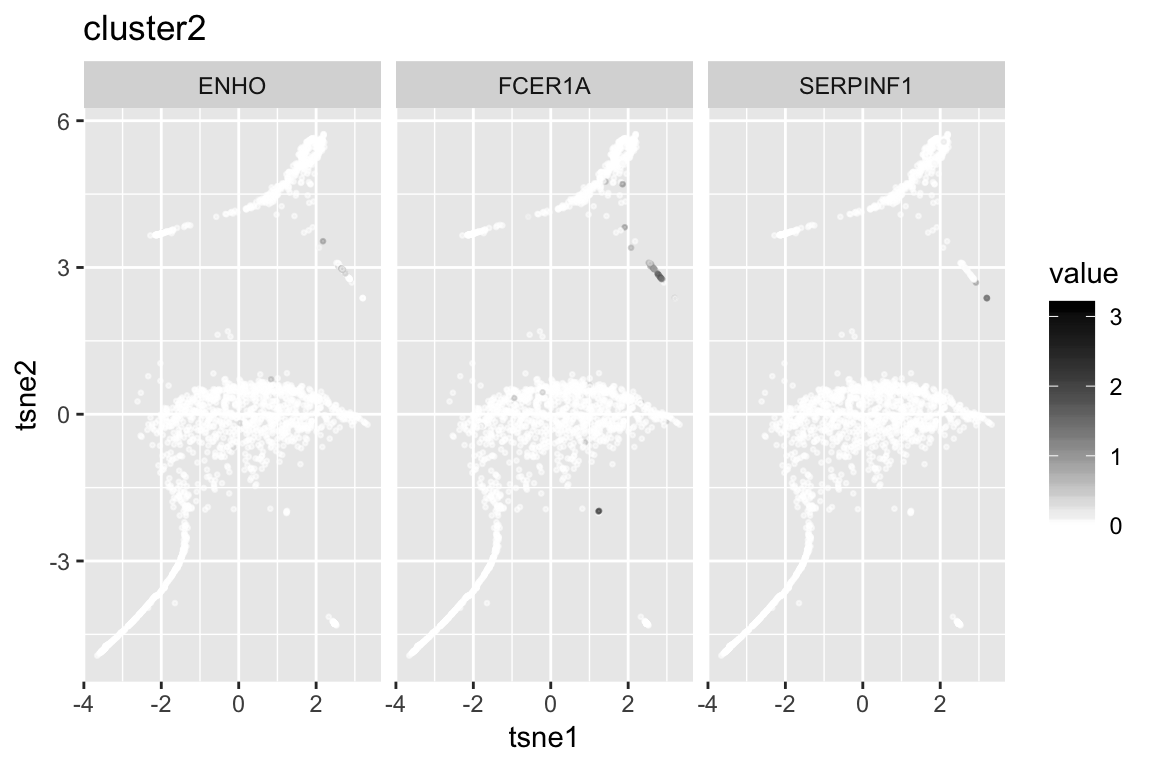
clust2_genenames clust2_log10p
1 SERPINF1 152.41836
2 FCER1A 143.19380
3 ENHO 79.10624
4 CLEC4C 77.80362
5 LRRC26 52.64777
6 SCT 52.64777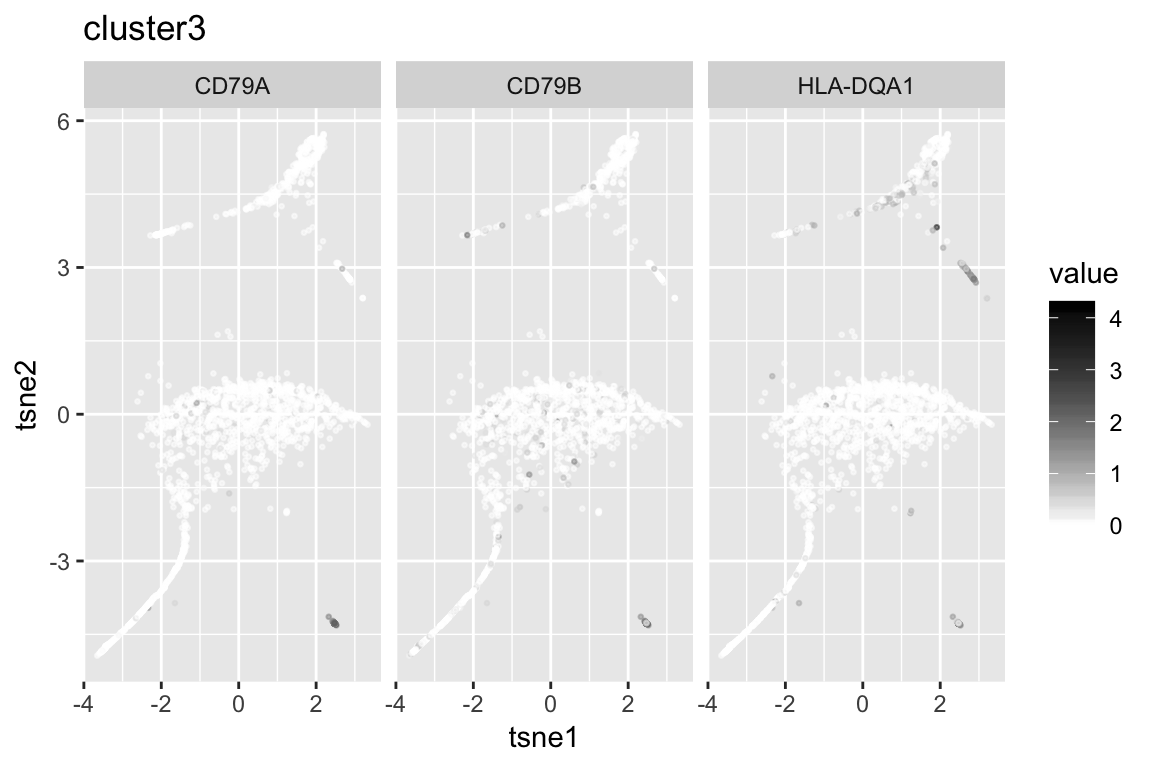
clust3_genenames clust3_log10p
1 CD79A Inf
2 HLA-DQA1 181.1766
3 CD79B 178.5284
4 HLA-DQB1 147.7264
5 FCRLA 134.5673
6 CD74 105.0767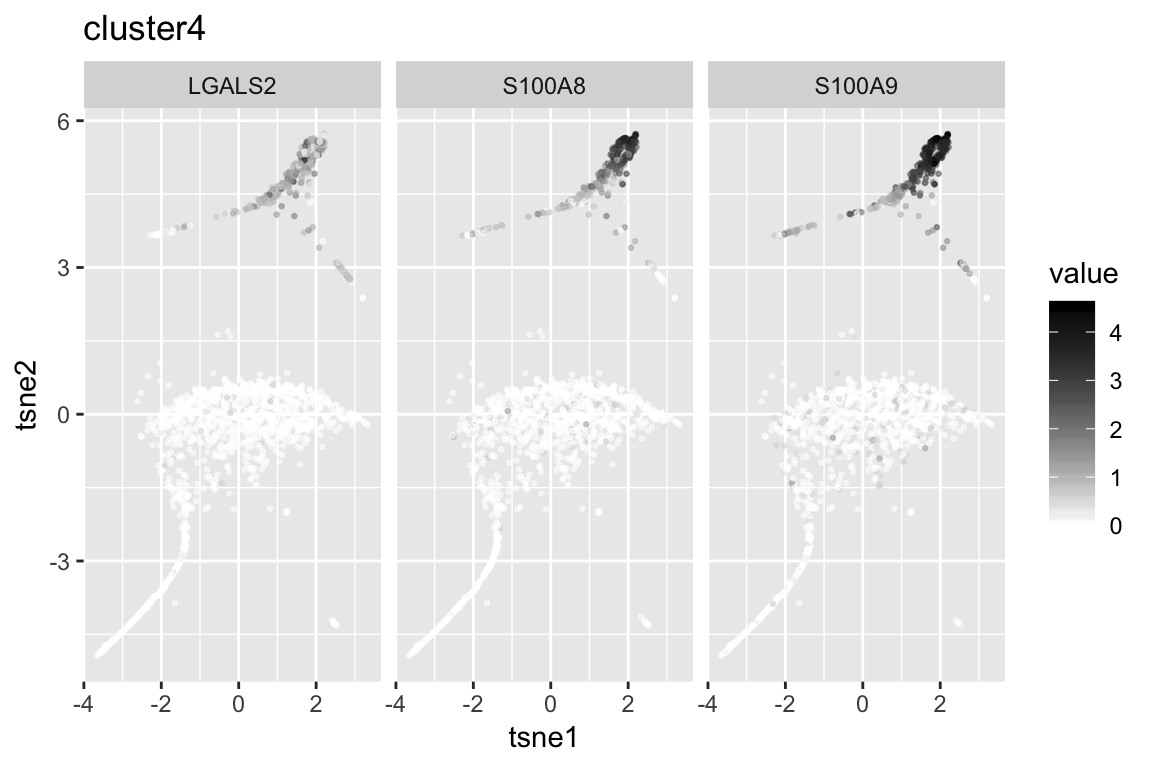
clust4_genenames clust4_log10p
1 LGALS2 Inf
2 S100A8 297.9284
3 S100A9 259.9251
4 FCN1 250.8238
5 LYZ 207.8968
6 CST3 204.0437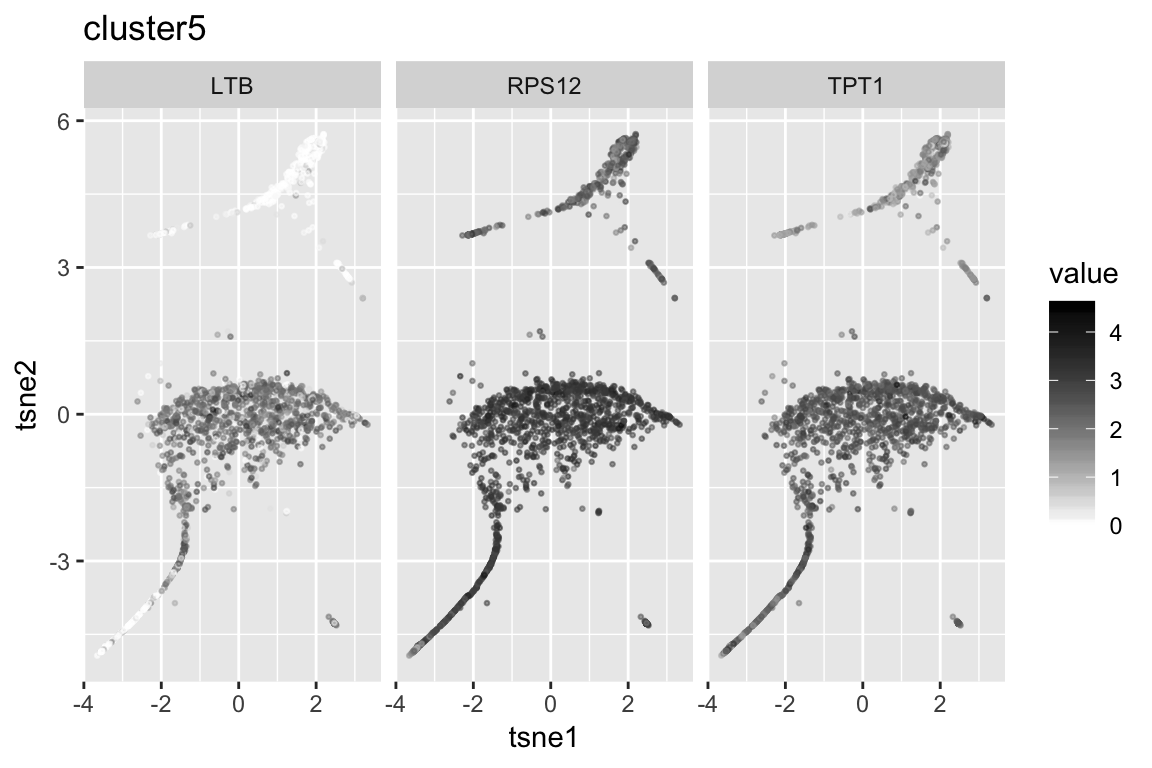
clust5_genenames clust5_log10p
1 TPT1 203.0014
2 LTB 202.5040
3 RPS12 192.9751
4 RPS27A 165.8921
5 RPL10A 164.1815
6 RPS5 154.5735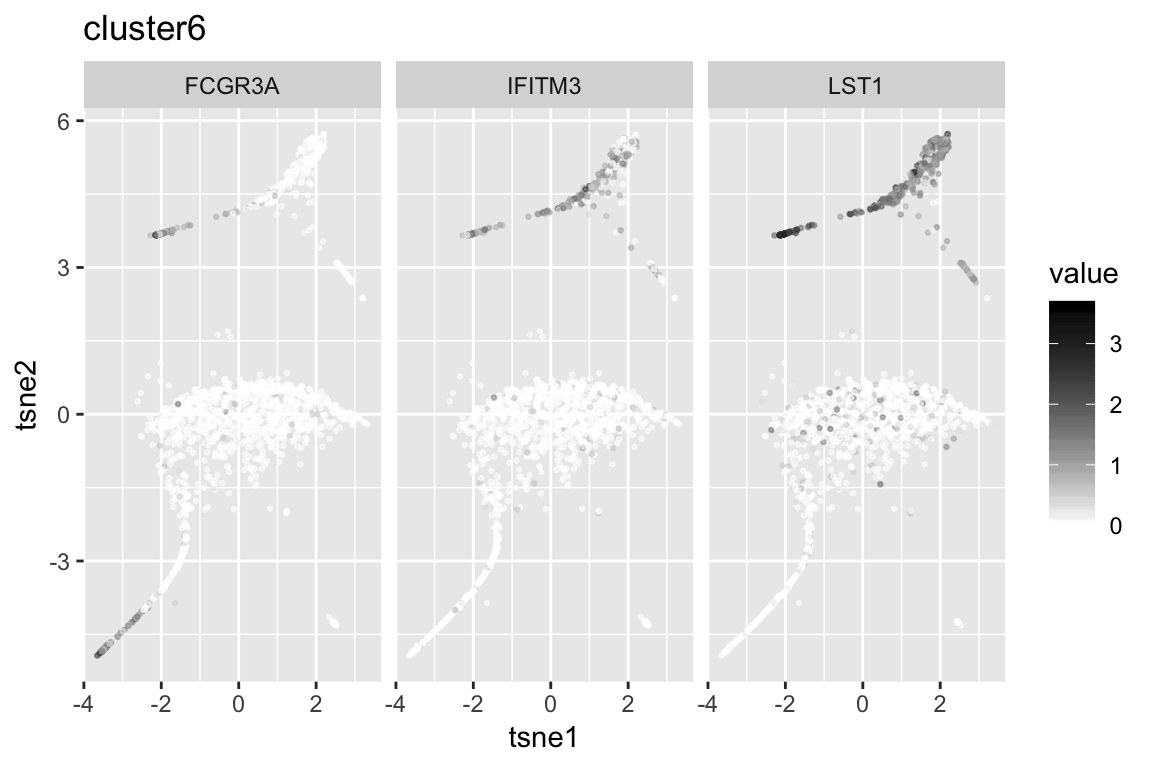
clust6_genenames clust6_log10p
1 FCGR3A 144.34980
2 IFITM3 124.06185
3 LST1 118.87115
4 AIF1 107.61418
5 FCER1G 107.49510
6 C5AR1 77.95185Session information
sessionInfo()R version 3.5.1 (2018-07-02)
Platform: x86_64-apple-darwin15.6.0 (64-bit)
Running under: macOS Sierra 10.12.5
Matrix products: default
BLAS: /Library/Frameworks/R.framework/Versions/3.5/Resources/lib/libRblas.0.dylib
LAPACK: /Library/Frameworks/R.framework/Versions/3.5/Resources/lib/libRlapack.dylib
locale:
[1] en_US.UTF-8/en_US.UTF-8/en_US.UTF-8/C/en_US.UTF-8/en_US.UTF-8
attached base packages:
[1] parallel stats4 stats graphics grDevices utils datasets
[8] methods base
other attached packages:
[1] bindrcpp_0.2.2 gridExtra_2.3
[3] SC3_1.8.0 SingleCellExperiment_1.2.0
[5] SummarizedExperiment_1.10.1 DelayedArray_0.6.2
[7] BiocParallel_1.14.2 Biobase_2.40.0
[9] GenomicRanges_1.32.6 GenomeInfoDb_1.16.0
[11] IRanges_2.14.10 S4Vectors_0.18.3
[13] BiocGenerics_0.26.0 SCNoisyClustering_0.1.0
[15] plotly_4.8.0 gplots_3.0.1
[17] diceR_0.5.1 Rtsne_0.13
[19] igraph_1.2.2 scatterplot3d_0.3-41
[21] pracma_2.1.4 fossil_0.3.7
[23] shapefiles_0.7 foreign_0.8-71
[25] maps_3.3.0 sp_1.3-1
[27] caret_6.0-80 lattice_0.20-35
[29] reshape_0.8.7 dplyr_0.7.6
[31] quadprog_1.5-5 inline_0.3.15
[33] matrixStats_0.54.0 irlba_2.3.2
[35] Matrix_1.2-14 plyr_1.8.4
[37] ggplot2_3.0.0 MultiAssayExperiment_1.6.0
loaded via a namespace (and not attached):
[1] backports_1.1.2 workflowr_1.1.1
[3] lazyeval_0.2.1 splines_3.5.1
[5] digest_0.6.15 foreach_1.4.4
[7] htmltools_0.3.6 gdata_2.18.0
[9] magrittr_1.5 cluster_2.0.7-1
[11] doParallel_1.0.11 ROCR_1.0-7
[13] sfsmisc_1.1-2 recipes_0.1.3
[15] gower_0.1.2 dimRed_0.1.0
[17] R.utils_2.6.0 colorspace_1.3-2
[19] rrcov_1.4-4 WriteXLS_4.0.0
[21] crayon_1.3.4 RCurl_1.95-4.11
[23] jsonlite_1.5 RcppArmadillo_0.8.600.0.0
[25] bindr_0.1.1 survival_2.42-6
[27] iterators_1.0.10 glue_1.3.0
[29] DRR_0.0.3 registry_0.5
[31] gtable_0.2.0 ipred_0.9-6
[33] zlibbioc_1.26.0 XVector_0.20.0
[35] kernlab_0.9-26 ddalpha_1.3.4
[37] DEoptimR_1.0-8 abind_1.4-5
[39] scales_0.5.0 mvtnorm_1.0-8
[41] pheatmap_1.0.10 rngtools_1.3.1
[43] bibtex_0.4.2 Rcpp_0.12.18
[45] viridisLite_0.3.0 xtable_1.8-2
[47] magic_1.5-8 mclust_5.4.1
[49] lava_1.6.2 prodlim_2018.04.18
[51] htmlwidgets_1.2 httr_1.3.1
[53] RColorBrewer_1.1-2 pkgconfig_2.0.1
[55] R.methodsS3_1.7.1 nnet_7.3-12
[57] labeling_0.3 later_0.7.3
[59] tidyselect_0.2.4 rlang_0.2.1
[61] reshape2_1.4.3 munsell_0.5.0
[63] tools_3.5.1 pls_2.6-0
[65] broom_0.5.0 evaluate_0.11
[67] geometry_0.3-6 stringr_1.3.1
[69] yaml_2.2.0 ModelMetrics_1.1.0
[71] knitr_1.20 robustbase_0.93-2
[73] caTools_1.17.1.1 purrr_0.2.5
[75] nlme_3.1-137 doRNG_1.7.1
[77] mime_0.5 whisker_0.3-2
[79] R.oo_1.22.0 RcppRoll_0.3.0
[81] compiler_3.5.1 e1071_1.7-0
[83] tibble_1.4.2 pcaPP_1.9-73
[85] stringi_1.2.4 pillar_1.3.0
[87] data.table_1.11.4 bitops_1.0-6
[89] httpuv_1.4.5 R6_2.2.2
[91] promises_1.0.1 KernSmooth_2.23-15
[93] codetools_0.2-15 MASS_7.3-50
[95] gtools_3.8.1 assertthat_0.2.0
[97] CVST_0.2-2 pkgmaker_0.27
[99] rprojroot_1.3-2 withr_2.1.2
[101] GenomeInfoDbData_1.1.0 grid_3.5.1
[103] rpart_4.1-13 timeDate_3043.102
[105] tidyr_0.8.1 class_7.3-14
[107] rmarkdown_1.10 git2r_0.23.0
[109] shiny_1.1.0 lubridate_1.7.4 This reproducible R Markdown analysis was created with workflowr 1.1.1